ClinGen Allele Registry (CAR) Performance and Guidelines¶
Registration Metrics - Higher Level Summary¶
| Registration rate / second |
Sort status |
Genomic Range |
Annotations in VCF? |
Return payload |
| 90k |
sorted |
narrow (<50M nt) |
N |
None |
| 20k |
sorted |
medium (50M < x < ~100M nt) |
N |
None |
| 5k |
unsorted |
wide (>~100M nt) |
N |
None |
| ~1k |
sorted / unsorted |
narrow / medium / wide range |
N |
Full |
| 100-900 |
sorted / unsorted |
narrow / medium / wide range |
Y |
Full |
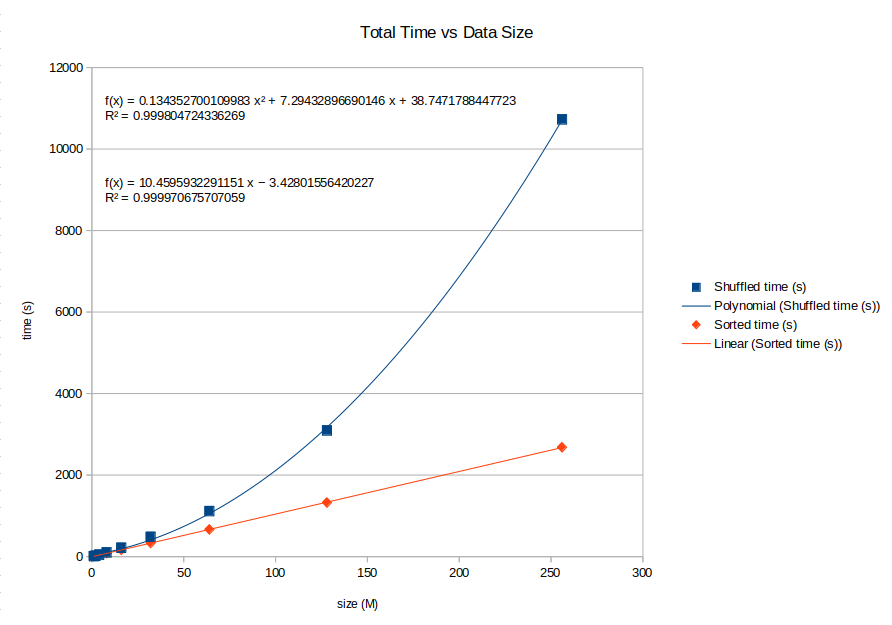
You will note that we observe increased performance (bottom to top):
- 4X increase in throughput by removing annotations (specific to VCF)
- 4X increase in throughput by returning no fields during registration
- 4X increase in throughput by sorting and utilizing a medium range of genomic location distribution
- 4X increase in throughput by sorting and splitting by narrow genomic ranges
High Volume Guidance¶
The Allele Registry provides for multiple routes for the bulk registration or bulk querying of variants. There are multiple factors that affect the turnaround time of an API request (can vary by 100X between API requests), namely:
- Network
- The size of the the submitted input, size of generated output, and network speed all factor in to the total request time
- Input size should be minimized when possible (e.g. removing annotation fields in VCF files)
- VCF_gnomAD shows the dramatic effect (80X) of removing the INFO field data prior to VCF registration / annotation
- Output size can be minimized by requesting only required JSON fields when possible
- fields=all returns full variant payload
- fields=none returns blank variant payload, but can be useful for verifying all input lines were processed (input lines == output lines)
- fields=none+@id returns only the CA ID of the variant
- API documentation has more information related to possible values to return
- Number of input entities
- The job time is also correlated with number of entities in the input
- <1M entries per API request is generally suitable. Heavily annotated VCF files input chunks should be decreased (25%, 50%, etc.) in the event of network timeouts.
- Ordering of entities
- Sorted input is not required, but very strongly advised! Sorted input requires loading less pages into RAM and dramatically decreases turnaround time.
- Distribution of entities across the genome
- Tighter distribution of input across chromosomes and chromosomal coordinates also requires loading less pages into RAM, which also decreases the total job time
- When possible, multiple datasets should be combined and sorted for large scale bulk registration
Common Input Types¶
I have HGVS input, where should I start?¶
- Obtaining_dbSNP_identifiers_for_CA_IDs - useful information showing how to process HGVS data from dbSNP, which can be applied to any HGVS data
- Sort your input by accession and starting coordinate
- You may find that splitting your input by reference sequence (especially for genomic variants) makes sorting easier
- Split your input up into multiple 'chunks' < 1M lines each
- Query the Allele Registry
- ./request_with_payload.sh "http://reg.clinicalgenome.org/alleles.json?file=hgvs&fields=none+@id" {input_file_name} > {output_file_name}.json
- Register variants in the Allele Registry
- ./request_with_payload.sh "http://reg.clinicalgenome.org/alleles.json?file=hgvs&fields=none+@id" {input_file_name} {user_name} {password} > {output_file_name}.json
I have VCF input, where should I start?¶
- VCF_gnomAD shows how gnomAD VCF data can be utilized
- Sort input based on chromosome and start coordinate
- Split your input into 1M chunks (see above regarding decreasing this number if you experience client timeouts)
- Note: per the above VCF gnomAD tutorial, you will need to insert the VCF header onto each split file
- Query the Allele Registry - return CA IDs
- ./request_with_payload.sh "https://reg.clinicalgenome.org/alleles?file=vcf&fields=none+@id" {input_file_name} > {output_file_name}.json
- Register variants in the Allele Registry - return CA IDs
- ./request_with_payload.sh "https://reg.clinicalgenome.org/alleles?file=vcf&fields=none+@id" {input_file_name} {user_name} {password} > {output_file_name}.json
- Annotate VCF - return CA IDs
- ./request_with_payload.sh ""http://reg.clinicalgenome.org/annotateVcf?assembly=GRCh38&ids=CA" {input_file_name} > {output_file_name}.vcf
- Annotate VCF and register variants - return CA IDs
- ./request_with_payload.sh ""http://reg.clinicalgenome.org/annotateVcf?assembly=GRCh38&ids=CA" {input_file_name} {user_name} {password} > {output_file_name}.vcf
- Header files
Registration Metrics - More Detailed¶
| Row |
Input Format |
CAR version |
Input characteristics |
Return type |
Database state |
Input size |
Output size |
Average registrations / second |
| A |
VCF |
64 |
unsorted, distributed across the full genome |
register only |
empty |
25MB |
3.7MB |
89,484 |
| B |
VCF |
64 |
sorted narrow distribution |
register only |
empty |
25MB |
3.7MB |
103,120 |
| C |
VCF |
64 |
unsorted, distributed across the full genome |
register, full payload |
empty |
25MB |
7.4GB |
2,805 |
| D |
VCF |
64 |
sorted narrow distribution |
register, full payload |
empty |
25MB |
9.9GB |
2,278 |
| E |
VCF |
64 |
unsorted, distributed across the full genome |
register only |
2.5B |
25MB |
3.7MB |
5,346 |
| F |
VCF |
64 |
sorted narrow distribution |
register only |
2.5B |
25MB |
3.7MB |
90,679 |
| G |
HGVS |
32 |
sorted narrow distribution |
query only |
2.5B |
38MB |
54MB |
22,222 |
| H |
VCF |
32 |
sorted, chr21, no annotations in input |
query, annotate VCF |
2.5B |
2.3MB |
. |
40,000 |
| I |
VCF |
32 |
sorted, chr21, full annotations in input |
query, annotate VCF |
2.5B |
1.4GB |
. |
500 |
Additional Notes¶
There are four main external factors related to CAR performance
- Network latency (input, output file sizes)
- The Allele Registry is cloud hosted by Rackspace and performance can be adversely affected by connectivity to their distributed servers, which is very rare.
- Additionally, the size of the input that needs to be uploaded and the size of the output that needs to be downloaded is a product of network latency.
- For example:
- Rows C vs. D we have an increased turnaround time for sorted, narrowly distributed input, which is correlated with the larger return size.
- Rows H vs. I we see a dramatic decrease in performance by including unnecessary input not required by the CAR (annotation details in the VCF INFO column)
- Number of input entities
- Performance scales linearly with number of input items. We generally recommend batches of < 1M per API query.
- Ordering of entities
- Submitting input in sorted order is highly encouraged and directly affects performance. This is due to loading pages of the CAR database into memory based on genomic location, thus variants in close proximity will be loaded into RAM for quicker access and minimizes the number of pages that need to be loaded.
- Distribution of entities across the genome
- Similar to the above, the narrower the window of input entities across the genome, the better the performance (similar to ordering entities mentioned above).
- For example, you can see the effect of unordered distributed entities across the genome adversely affect the performance (A vs. B, E vs. F)
Notes:
- API documentation:
- The current version of Allele Registry as of 6/3/22 is utilizing 32 bit infrastructure. We are planning on rolling out 64 bit in the near future, which among other things will provide support for registration of > 4.2B canonical alleles (currently at ~2.57B).
- The sorted narrow distribution data referred to in rows B, D, and F has a range of ~10M nt and the input in row G has a range of ~20M
- Obtain CA IDs for HGVS (row G) tutorial:
- Annotate VCF (rows H, I) tutorial
- Database growth
- Another function of database growth is initial increased turnaround time (A-D, vs. E-I). Again, this is related to the number of pages that are required for loading into memory and we will continue to monitor performance as we increase the number of registered alleles to 3B, 4B, etc.
CAR Performance - Sorted vs. Unsorted¶